What are the advantages of barcoding in mass cytometry? In short, time and money for drug developers.
Here’s an example with 120 specimens. Let’s compare what happens without barcoding and with barcoding.
| Parameter | Without barcoding | With barcoding |
| Processing method | Individual processing per specimen | Samples barcoded and pooled |
| Number of runs | 120 runs (1 per specimen) | 6 runs (20 specimens per run) |
| Data Quality | Variable | Consistent and reliable; reduced cell loss |
How does barcoding work? Read on to find out.
Example of barcoding in mass cytometry on 120 specimen project
To illustrate how barcoding works in mass cytometry, let’s consider a simple example on the 120 specimen project using metal isotopes for tagging samples.
Goal: Barcode 20 different samples for simultaneous analysis. Remember, we had a 120 specimen project, with 20 specimens in each of the six runs.
First, the barcode criteria must be established:
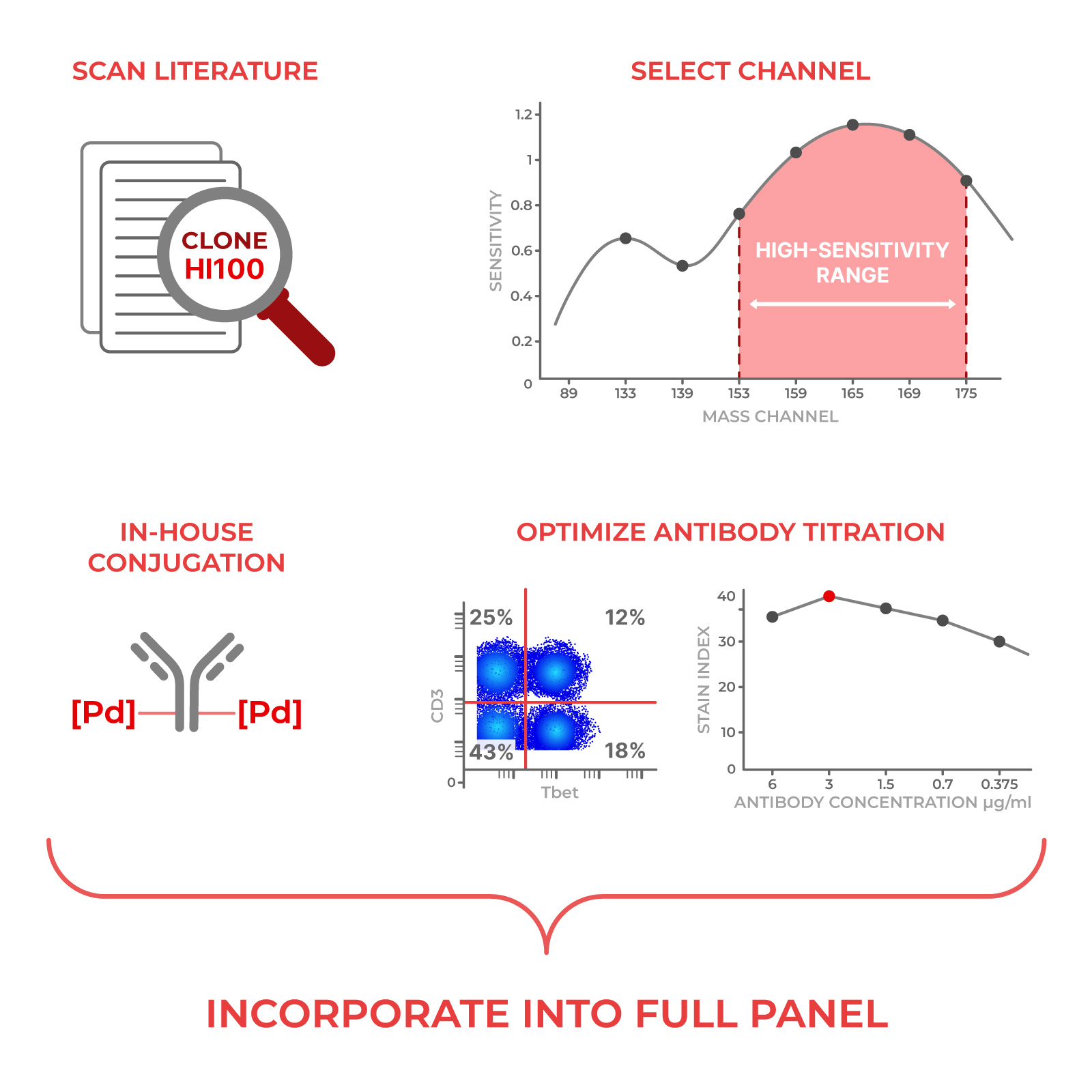
Purity: each isotope must be >90% pure
Titration: each isotope is titrated to the concentration that gives the best signal-to-noise ratio. This is similar to what we do with antibodies. We also ensure each isotope produces similar “brightness.”
Combination: after each isotope is titrated, combine three isotopes together into one barcode and test again the optimal concentration to be stained with cells
Second, we pick the barcoding isotopes: We’ll use six palladium isotopes not used in the 40+ marker antibody panel. To construct the actual barcode, we’ll pick three from those six, to avoid signal overlap between the channels.
¹⁰²Pd
¹⁰⁴Pd
¹⁰⁵Pd
106Pd
108Pd
110Pd
Now, the fun part starts.
Third, we build a barcoding scheme:
By using different combinations of these three isotopes, we can create unique barcodes for each sample. Here’s an example:
| Mass in Angstroms | ||||||
| Specimen ID | 102 | 104 | 105 | 106 | 108 | 110 |
| 01 | 1 | 1 | 1 | 0 | 0 | 0 |
| 02 | 1 | 1 | 0 | 1 | 0 | 0 |
| 03 | 1 | 1 | 0 | 0 | 1 | 0 |
| 04 | 1 | 1 | 0 | 0 | 0 | 1 |
| 05 | 1 | 0 | 1 | 1 | 0 | 0 |
| 06 | 1 | 0 | 1 | 0 | 1 | 0 |
| 07 | 1 | 0 | 1 | 0 | 0 | 1 |
| 08 | 1 | 0 | 0 | 1 | 1 | 0 |
| 09 | 1 | 0 | 0 | 1 | 0 | 1 |
| 10 | 1 | 0 | 0 | 0 | 1 | 1 |
| 11 | 0 | 1 | 1 | 1 | 0 | 0 |
| 12 | 0 | 1 | 1 | 0 | 1 | 0 |
| 13 | 0 | 1 | 1 | 0 | 0 | 1 |
| 14 | 0 | 1 | 0 | 1 | 1 | 0 |
| 15 | 0 | 1 | 0 | 1 | 0 | 1 |
| 16 | 0 | 1 | 0 | 0 | 1 | 1 |
| 17 | 0 | 0 | 1 | 1 | 1 | 0 |
| 18 | 0 | 0 | 1 | 1 | 0 | 1 |
| 19 | 0 | 0 | 1 | 0 | 1 | 1 |
| 20 | 0 | 0 | 0 | 1 | 1 | 1 |
Fourth, onto the actual processing steps in the lab:
- Barcode individual samples:
Each of the 20 samples is incubated separately with a specific combination of the barcoding isotopes from the table above.
The isotopes bind to proteins uniformly across all cell types in the sample. - Pool samples:
After barcoding, all 20 samples are combined into a single tube.
The pooled sample is then stained with a panel of metal-tagged antibodies targeting specific cellular markers (using different isotopes not overlapping with the barcodes). - Acquire the data:
The pooled, stained sample is run through the mass cytometer.
The cytometer detects the metal isotopes from both the barcodes and the antibody-bound markers. - Deconvolve the data:
Specialized software analyzes the data, identifying cells based on the presence of barcoding isotopes.
Cells are assigned back to their original samples according to the unique combination of isotopes detected.
For example, a cell positive for 102Pd, 104Pd for 110Pd is assigned to Specimen 04. - And then, analyze!
Once deconvoluted, each sample’s data can be analyzed individually for immunophenotyping.
This includes assessing the expression of various markers to characterize cell populations.
So what just happened?
A drug developer who sends 120 specimens, can get them done in 6 runs, not 120. That means lower batch variation, and better quality data for your patients.
Bonus: what if we need more barcodes?
You might have noticed that we used 6 isotopes to generate barcodes, which yields 20 combinations (6 Choose 3). As the number of specimens per run increases, you can add more isotopes to generate higher combinations (i.e. 10 isotopes can generate 120 combinations).