“I have a 13 marker panel that analyzes T-cells. And my drug just hit the T-cells, why bother looking at any other immune cell types, like B cells, or NK cells? Even if those other cells are affected, I don’t know how to interpret those results.”
The reason is: those extra subsets might predict the difference between success and failure for a drug.
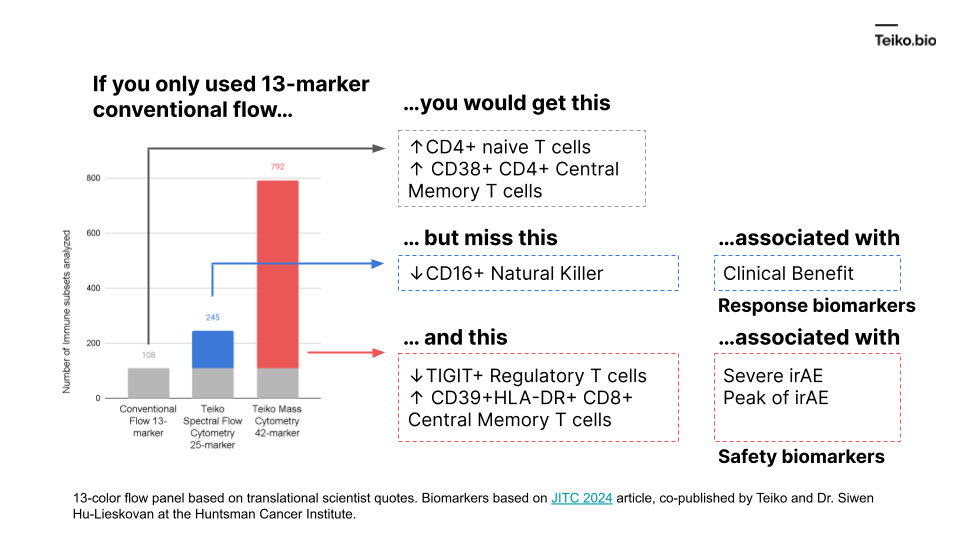
More to the point, if we took a conventional flow panel, you get up to ~108 subsets with a 13-marker panel, almost exclusively T cells. Using mass cytometry, with a 42-marker panel, you get to 792. With spectral flow, it’s around 245. This diagram shows what is missed by the lower-parameter methods. And these are cell subsets we ourselves have found in our own work and collaborations, associated with either clinical benefit, severe immune-related adverse events or peak toxicity.
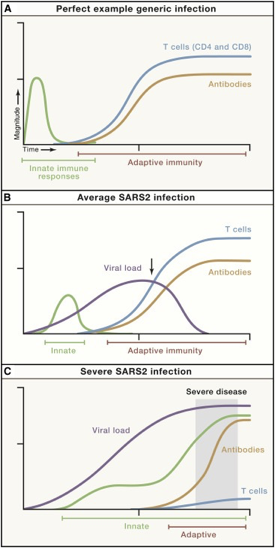
We know there is a “characteristic” time-series curve associated with clearing infections, like COVID. It would be difficult for a drug developer to analyze a “productive” immune response if whole curves were missing. For example, imagine tracking one curve, like the antibodies. If you tracked just antibodies, you could be misled into thinking someone had an average COVID infection or generic infection.
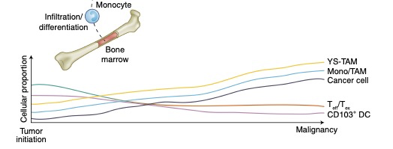
There is good reason to believe this sort of curve is true in cancer immunology. To that point, this is a nice time-series diagram from Nature Medicine of different cell types in the tumor microenvironment. The authors summarized it well: “understanding what cell types can be modulated and when may enable the next biggest improvements in immunotherapy.”
These distinct subtypes are not of academic interest: these drive real-world responses.
Pre-fixation versus post-fixation. Sounds complicated. What do those terms mean, anyway?
Pre-fixation: The pre-fixation time is from drawing blood to putting it into our first buffer “stable lyse,” starting the blood fixation process. This stage needs to be done within five hours from the time of blood draw.
(The time between adding Buffer 1 and adding Buffer 2 is the “fixation time” which is a separate measurement that should be the recommended 15 min. At this point, the sample is “fixed”: meaning the cells have been “snapshotted” in place.)
Drug developers care about this measure because it can tell them how long a phlebotomist has to put the blood sample into a collection kit. A five hour window gives drug developers the flexibility to collect blood samples from multiple patients throughout the morning and process them all in the afternoon, or collect in the afternoon and process at the end of the day.
Post-fixation: The amount of time once the sample has been fixed and frozen, to when the sample is actually run on the cytometer and data is produced. This can be on the order of months, or even years.
Drug developers care about this measure because it can tell them how long the sample is stable for. For trials that run for 24 months, drug developers want to know that they can get the same quality of data from the 24 month-old samples from their first patient as their most recent patient.
The answer depends on the confidence you need, and the variability you can accept. Variability is expressed by the coefficient of variation (CV) is simply the standard deviation divided by the mean. The higher this number, the more “variable” the measurement. The lower the number, the less “variable” the measurement. Intuitively, a population that appears a lot, say 10% of the time, needs fewer cells than a population that occurs 0.001% of the time.
For purposes of this answer, let’s consider two populations, intermediate Monocytes (inMono) and T cells. These figures are derived from our mass cytometry validation report: https://teiko.bio/technology/, however we believe these reference ranges are applicable regardless of the instrument type (spectral cytometry or mass cytometry).
Cell Population
Median
Inter-run CV%
Intra-run CV%
inMono
0.47%
16.09%
4.63%
T cells
37.28%
0.39%
1.27%
Table 1: Reference Range Values for PBMCs from Healthy Subjects, % of non-granulocytes
Luckily, this question has been addressed in the work of Keeney et al. To wit, “for cell-based assays such as flow cytometry, a simple calculation can be used to determine the size of the database/sample that will provide a given precision: r = (100/CV)2; where r is the number of events meeting the required criterion, and CV is the coefficient of variation of a known positive control.”
We’ve adapted Keeney’s table below:
Desired Coefficient of Variation (%)
1
5
10
20
r = number of events of interest
10,000
400
100
25
When occurring at a frequency of:
Fraction
1:n
Total number of events which must be collected
0.1
10
100,000
4,000
1,000
250
0.01
100
1,000,000
40,000
10,000
2,500
0.001
1,000
10,000,000
400,000
100,000
25,000
0.0001
10,000
100,000,000
4,000,000
1,000,000
250,000
0.00001
100,000
1,000,000,000
40,000,000
10,000,000
2,500,000
0.000001
1,000,000
10,000,000,000
400,000,000
100,000,000
25,000,000
Table 2: “Determination of database/sample size that will provide a given precision in rare event analysis”, Keeney, et. al
Now, let’s couple that with the instruments we have at our disposal: mass cytometry and spectral flow cytometry. Based on our experience, mass cytometry and spectral flow have recovery rates of 50% and 90%, respectively.
Table 3: Estimated events for Peripheral Blood Mononuclear Cell collection
Drawn from Patient (mL)
Cells per mL (M)
Number of Cells in Whole Blood (M)
% Granulocytes
Approximate number of Granulocytes (M)
Non-Granulocytes (M)
Instrument
Recovery Rate
Resulting Events (M)
Resulting non-granulocyte events (M)
2
5
10
50%
5
5
Mass Cytometry
50%
5.00
2.50
5
5
Spectral Flow
90%
9.00
4.50
Table 4: Estimated events for Whole Blood collection
For PBMCs, you can get 0.5M – 10.8M events, depending on the volume of blood collected. In the table, we just show the average ranges, i.e. 2.63M – 4.73M. And for whole blood, you can get 4M – 10.8M total events. Since at least 50% of the cells will be granulocytes, you’ll get to ~2M – ~5.4M non-granulocytes. For readability, we’re showing only a smaller range, from 2.5M – 4.5M.
Now, let’s come back to the two populations from Table 1, inMono and T-cells.
Desired CV
1%
5%
Teiko observed inMono Median % of non-Granulocytes
0.47%
Total Number of events that must be collected, based on Keeney Table
1,000,000
40,000
Estimated inMono Population needed to achieve CV
4,700
188
Actual Teiko collected inMono events
302
Above Threshold?
No
Yes
Actual Teiko Intra-Run CV
4.63%
Actual Teiko Inter-Run CV
16.09%
Industry Standard CV Acceptance Criteria
25-30%
Table 5: Desired CV for inMono
For a 5% CV for a 0.47% population, you would need about 188 events to achieve this CV. And We collected 302, so we were above the threshold. Turns out, we were right in line with inter-run CVs, at 4.63%, and Inter-run CVs at 16.09%.
Desired CV
1%
5%
Teiko observed T-cell Median % of non-Granulocytes
37.28%
Total Number of events that must be collected, based on Keeney Table
100,000
4,000
Estimated T-cell Population needed to achieve CV
37,280
1,491
Actual Teiko collected T-cell events
39,210
Above Threshold?
Yes
Yes
Actual Teiko Intra-Run CV
1.27%
Actual Teiko Inter-Run CV
0.39%
Industry Standard CV Acceptance Criteria
25-30%
Table 6: Desired CV for T-cells
Now, T-cells are much more populous. In our precision study, we collected 39,210 events, beyond the 37,280 and 1,491 cells we need to achieve a good coefficient of variation. And, as it turns out, we clocked 1.27% CVs for intra-run and 0.39% CVs for inter-run.
So, hopefully this gives you an intuition for “How many cells do you need to reliably detect a population of interest?” Interested in capturing a specific population?
In short: We validate antibodies by testing different antibody clones, concentrations, and cellular conditions to optimize signal-to-noise ratio and reduce background noise, ensuring accurate identification of cellular markers. For the full details, download our technical whitepaper ‘Panel and Custom Antibody Validation Process‘ to learn more!
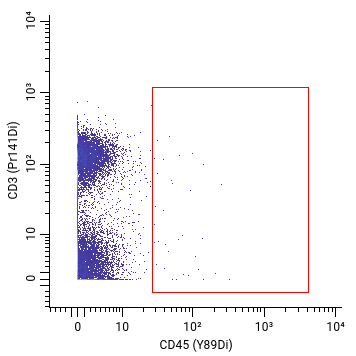
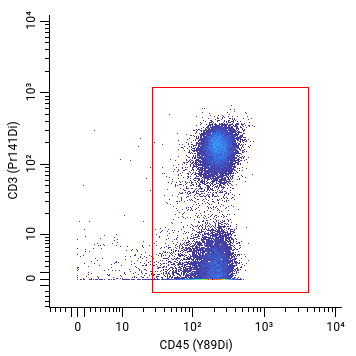
Our customers often want to know that when we have a cellular marker on our panel, such as CD45, we can confidently distinguish this cellular marker signal from background noise. To detect a cellular marker, we need a corresponding clone.
But not all clones are created equal. For instance, here’s an example of two different CD45 antibody clones tested on human cells with very different results. Can you see the difference?
Bad signal: cellular population missing or too low
Good signal: cellular population found
To that end, our immunologists use their extensive expertise to reduce mass cytometry panel validation time from months to weeks, using proven techniques.
Problems with standard approaches: Traditional approaches would use panel design software alone to assign markers to channels, however standard panel design software approaches have crucial limitations. Specifically, these approaches:
only work with markers sold by the manufacturer
leave the antibody clone, antibody concentration, and stimulation condition unspecified for markers not sold by the manufacturer
Imagine you need a marker that’s not sold by the manufacturer: at this point, you need an expert to figure out what the right clone and concentration is. And if you don’t happen to have an expert handy, and you get the wrong clone or concentration: that could yield “bad or missing signals” on a panel, and ultimately unuseable data for precious samples.
Now, let’s compare our approach.
Our process:
We pair the best of software design with expert scientist intuition. Teiko scientists and immunologists have collectively processed over 2,800+ samples and 25 panels, yielding many hard-won lessons about panel design.
Ultimately, for a 44-marker panel, the goal of this process is to produce a 44 X 5 table:
Marker
Channel [44 total]
Antibody Clone
Antibody Concentration (μg/ml)
Cellular conditions for signal detection
CD45
[89Y, 112Cd, …, 209Bi]
[C1, C2, … CN]
[6, 3, 1.5, … 0.1875]
[Stimulated PMA, Stimulated PHA, Unstimulated]
CD8a
[89Y, 112Cd, …, 209Bi]
[C1, C2, … CN]
[6, 3, 1.5, … 0.1875]
[Stimulated PMA, Stimulated PHA, Unstimulated]
…
…
…
…
…
CD45RA
[89Y, 112Cd, …, 209Bi]
[C1, C2, … CN]
[6, 3, 1.5, … 0.1875]
[Stimulated PMA, Stimulated PHA, Unstimulated]
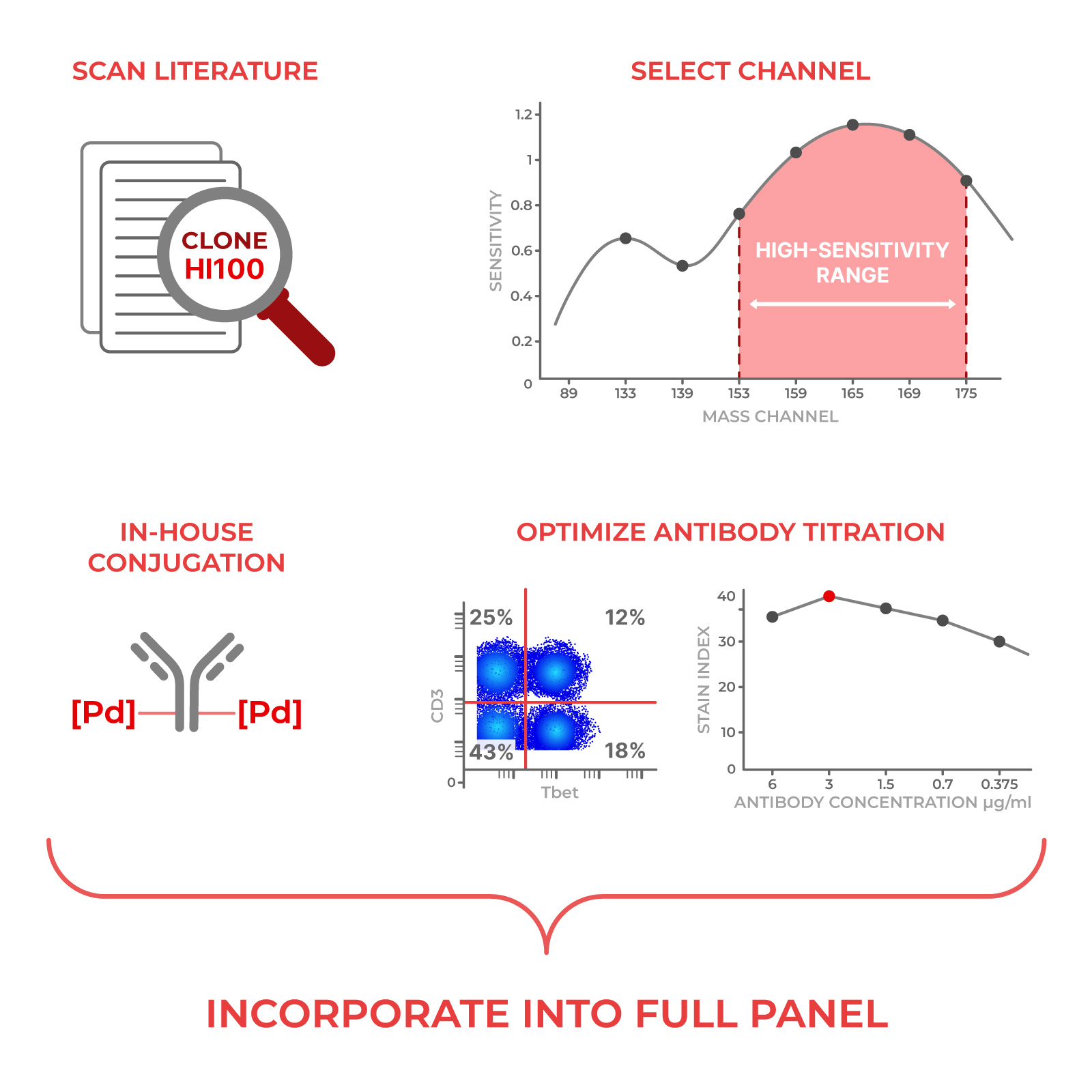
The first thing we do is look at the literature and antibody vendors and see if a marker of interest has been tested before. For each marker, i.e. CD45, we’ll figure out which antibody clone (i.e. HI30) to use based on historical performance.
Then, we’ll determine which channel to use to get the “maximum” signal to noise ratio. In general, Teiko’s experts follow a few rules that sound simple but require a lot of knowledge to execute quickly:
If a marker is weakly expressed on the cells, we put that marker on a “bright” channel to get a good signal.
Similarly, if a marker is strongly expressed on the cells, we put that marker on a “dim” channel to prevent the signal from becoming too “overwhelming”.
Some metals shouldn’t be matched on the same cell population because of concerns for natural impurity, and we won’t put those metals together.
Our experience with channel placement is where our experts can save you weeks of time and frustration.
After finding the right channel, we determine the “optimal” antibody concentration to get the maximum separation between signal and noise. We plot this “stain index” on a curve to find the right concentration.
This is how we generate the stain index:
First, we determine whether the marker needs to be tested in unstimulated or stimulated cells based on whether the marker is expressed on immune cells at baseline. If stimulation is required, we always test two different biological stimulations.
Next, we test six different concentrations of antibody and at each concentration we check:
Strength of the signal
Presence of background noise
“Spillover” (false-positive) signal into other channels
When a marker calls for it, we focus our analysis on a specific population that expresses that marker, so we don’t underestimate the signal.
Finally, we check that our marker performance is comparable to what others have seen in literature.
The end result is a fully populated table and completed validation:
Marker
Channel [44 total
Antibody clone
Antibody Concentration (μg/ml)
Cellular Conditions for Signal Detection
CD45
89Y
C2
1.5
Unstimulated
CD8A
209Bi
C1
0.1875
Unstimulated
…
…
…
…
…
CD45RA
112Cd
C3
3
Stimulated PMA
Illustrative purposes only
What is your process for validating new antibodies and adding them into my customized panel?
Using a process very similar to how we validated our base panels, we verify the optimal staining concentration at which each metal-conjugated antibody demonstrates a detectable and accurate signal on the mass cytometer, while minimizing background signal and spillover into other channels. Once titrated and validated individually, these antibodies are added to the panel and the full panel is then validated.
In sum
That’s a lot of heavy lifting behind the scenes. And we do that in weeks, not months. Teiko scientists performed this process to design all of our current Custom backbone and Standard panels.