The complementary capabilities of single-cell RNA seq and high-dimensional cytometry
Published by Teiko BioNovember 7, 2022 (7 min read)
Single-cell RNA sequencing (scRNA-seq) and high-dimensional cytometry (i.e. spectral flow cytometry or mass cytometry), are both popular approaches for high dimensional analysis of disease states with single cell resolution. Individually, each is a powerful tool for determining the mechanisms underlying disease development and assessing response to treatment. When combined, however, the two can deeply enrich mechanistic and biomarker therapeutic research by providing complementary breadth and depth of analysis.
Researchers have used scRNAseq and high-dimensional cytometry as complementary tools in a wide variety of published studies to provide additional breadth, depth, and coverage when analyzing cellular processes. Below are a few specific examples that illustrate the diversity and range of these approaches in a variety of biomedical research settings.
Finding Cells of Interest in the Tumor Microenvironment
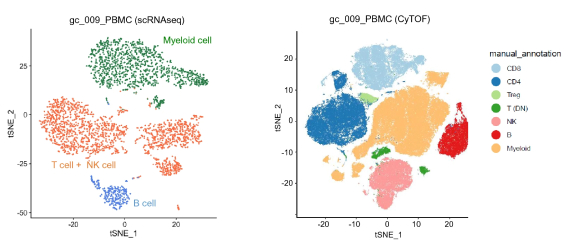
Comparison of scRNAseq data and CyTOF data taken from Figure 2a of Kashima et al. Sci Rep (2021) without alterations under the Creative Commons Attribution 4.0 International License. Full citation can be found under References below.
A study published in Scientific Reports by Kashima et al. in 2021 used both scRNA-seq and high-dimensional mass cytometry to investigate the immune profiles of the tumor microenvironment in gastric cancer patients.1 Because scRNA-seq was not limited by predetermined marker selection, it was able to detect and characterize new cell populations and subgroups within those populations. For example, scRNA-seq was able to identify plasma cells that were not defined by mass cytometry markers. In contrast, mass cytometry showed greater precision in differentiating predefined cell types characterized by a specific pattern of protein expression. For instance, NK cells and T cells were differentiated with greater accuracy using mass cytometry than with scRNA-seq.
As a result of their research, Kashimi et al. concluded that the greatest strength of high-dimensional mass cytometry lies in its ability to provide “narrow and clear” detection of prespecified cell types, while scRNA-seq enables “wide and indistinct” characterization of cells of interest. Thus, one approach for using the techniques together would be to apply scRNA-seq for discovery of new cell types of interest given the wide-breadth of this approach and then applying high-dimensional cytometry for more rapid and granular characterization of these cell types.
Brain Matters: Protein Provides the Persistent Signal
Fernández‐Zapata et al. published a review article in Brain Pathology in 2020 summarizing studies that made use of both high-dimensional mass cytometry and scRNA-seq to comprehensively analyze the structure and function of human microglia, the central nervous system’s native immune cells.3 ScRNA-seq was effectively utilized to characterize microglial subsets formed in response to CNS pathology, such as multiple sclerosis (MS).
A key point from the Fernández‐Zapata et al. article is that function is mediated at the protein level, and proteomic techniques like mass cytometry can directly measure protein expression, turnover, and post-translational modifications missed by scRNA-seq. For example, the authors discovered that “for some molecules with low transcript levels such as CX3CR1, CSF1R and FCGR1A, [there was] no change or even an increase in protein expression.” This means that for infrequently produced markers, measuring protein expression will provide a more accurate readout of the cell state. One should also be aware that there is also greater cellular “noise” in scRNA-seq since there are significant differences in gene expression from cell to cell at the level of mRNA transcription even in uniform cell populations.4 Transcription (mRNA production) occurs in pulses.5 ScRNA-seq captures a “snapshot” in the life of a cell that ebbs and flows in terms of transcription. Lack of detection of a particular mRNA does not mean the cell never produces it, just that the cell does not require it at that specific point in time. By contrast, proteins are longer-lived molecules allowing for a greater time span to detect them and to capture cell activity as cells transition from one state to another.
Nanoparticles are frequently added to consumer and biomedical products, and their interactions with human immune cells are important to characterize to ensure their safety. To address this topic, Ha et al. published a study in the journal Small in 2020 exploring the interactions between silver nanoparticles (AgNPs) and primary immune cells using both high-dimensional cytometry and scRNA-seq.2 Using mass cytometry, the study authors were able to show that monocytes and B cells had more contact with AgNPs than other cell populations. ScRNA-seq was then able to further characterize the specific cellular reactions in each cell type induced by contact with AgNPs.
In this study, mass cytometry was used as an initial screen for quick and accurate identification of cell subsets of interest that physically interacted with the specific nanoparticles being studied. It was able to directly detect the cell-AgNP interactions on a single cell level. ScRNA-seq was then applied to obtain additional transcriptomic characterization taking place within the different cell types downstream of the interaction. In this instance the researchers were interested in AgNP-cell interactions, however a similar approach could be used to detect physical interactions between immune cells and any heavy metal labeled molecule.
An additional consideration when applying scRNA-seq is to determine how “deep” or “shallow” the analysis should be, which will depend on time and budget constraints.1,6 A “shallow” analysis (low number of mRNAs read per cell relative to the total mRNA content of the cell) carries greater uncertainty about whether transcriptomic differences are due to biological processes or sampling variation. Deeper sequencing (reading more mRNA molecules) enables greater resolution in differentiating cells, but big-picture population dynamics can be lost. An initial “shallow” analysis is not necessarily a bad thing for early-stage exploration and mapping out broad networks and processes. Since mass cytometry is currently limited to around 45 parameters, it must cover a broad range of preselected protein markers that help to distinguish cell types amongst diverse populations. It is also constrained by the technical challenges involved in constructing marker-specific antibodies conjugated to metal-isotopes.1 On the flip side, the longer half-life of proteins means they go through fewer fluctuations in comparison to mRNA and thus are more stable and reliable cell markers.5 Furthermore, high-dimensional cytometry techniques like mass cytometry and spectral flow cytometry have higher throughput, with the ability to analyze hundreds of thousands or even millions of cells. Higher throughput translates to greater confidence that a complete picture of cell population changes is being captured.
Teiko Bio: A Natural Complement to scRNAseq
Whether using high-dimensional cytometry to identify large scale immune cell population changes, study subset interactions, explore specific subsets with greater depth and accuracy, or validate function at the protein level, high-dimensional cytometry with Teiko is a natural complement to scRNAseq. Together the two techniques can form a powerful duo to accomplish your clinical research goals.